Structural Levels of Proteins: Primary, Secondary, Tertiary & Quaternary Structure
This chapter discusses the four levels of protein structure, structural databases, and tools used in protein structure and function analysis.
Overview
To understand the fundamental principles of three-dimensional protein structure and its potential applications in academia and industry, we first need to understand the four levels of protein structure. These levels are interdependent, forming a complex network of interactions among hundreds or even thousands of protein atoms, often involving solvent molecules, various ligands, and metal atoms. This chapter will discuss the basic principles of protein structure and bioinformatics databases used to search for protein structure-related information.
Primary, Secondary, and Tertiary Structure
The first fundamental level of protein structure is the amino acid sequence, often called the primary structure. During protein synthesis, the 20 most common amino acids are linked to form a polypeptide chain, a process catalyzed by the ribosome. The amino acid sequence largely determines the secondary structure, which includes α-helices and β-sheets, and the tertiary structure, characterized by domains and specific folds. However, it is often essential to consider the influence of the local environment on secondary and tertiary structure stabilization. A clear example is membrane proteins, which are embedded in a hydrophobic lipid environment. These proteins’ secondary and tertiary structures depend on the membrane environment for stability. When removed from the membrane, they proteins lose their native structure (a process known as denaturation), aggregate, and precipitate from solution. Detergents are often used to preserve the three-dimensional conformation and ensure the proteins remain soluble and functional during purification. The detergent creates micelles that surround the hydrophobic parts of the protein, mimic the membrane lipids, and help stabilize the protein’s secondary and tertiary structure.
The spatial arrangement of secondary structure elements determines a protein’s tertiary structure. This specific arrangement is known as a “fold,” a unique characteristic of a protein’s structure. Currently, approximately 1,300 distinct folds have been classified in three-dimensional protein structures. In a later chapter, we will explore examples of these folds and the CATH database, where folds and domains are categorized. This classification helps to reveal the relationships between the folds and the evolutionary origins of proteins.
Motifs, Folds & Domains
A domain is the primary classification unit in proteins. It is an independent folding unit because it can often be cloned, expressed, and purified separately from other domains of a multi-domain protein while maintaining the same structural characteristics. It may even exhibit similar activities while present in different proteins. Such activities may include binding small molecule ligands or interacting with other proteins. Each protein domain is assigned a specific fold. Some proteins consist of a single domain, while others can contain two or more domains with different folds. Although domains with similar folds may not necessarily be related functionally or evolutionarily, the fold can still provide insight into the evolutionary origin of a protein.
In addition to domain conservation, there are other types of conserved structural elements in proteins called structural motifs. These smaller structural units may be present within different and not necessarily evolutionary-related domains. Examples of such motifs include helix-turn-helix motifs, β-hairpins, and the Greek key motif. However, these motifs are not considered independent folding units in contrast to domains. More detailed discussion and examples can be found on the domains, folds, and motifs page.
Quaternary Structure and Oligomers
The next level of protein structure is the quaternary structure, also known as oligomeric structure. This structure is formed by two or more polypeptide chains called subunits (not to be mixed with domains). An oligomer can consist of identical subunits, known as a homo-oligomer, or it can be composed of different protein molecules, referred to as a hetero-oligomer. Oligomeric structures serve various functions within cells and often act as molecular machines that utilize ATP (adenosine triphosphate) as an energy source. An example of an oligomeric structure is the enzyme Mg-chelatase, illustrated in the image on the right.
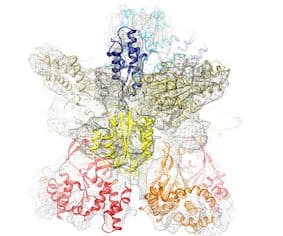
An oligomer is stabilized by interactions between its subunits, such as hydrophobic interactions, hydrogen bonds, and salt bridges. In cases of multi-subunit enzymes, the subunits within the structure often contribute to the formation of the active site or other ligand binding sites. An oligomeric complex may also interact with other proteins, forming a transient complex.
Magnesium chelatase, shown in the images above, is involved in chlorophyll biosynthesis. It catalyzes the insertion of magnesium into protoporphyrin IX, which is the first committed step in chlorophyll biosynthesis. About 20 more catalytic reactions are required before the chlorophyll molecule is synthesized. Mg-chelatase has three different subunits. In bacteriochlorophyll synthesis, they are named BchI, BchD, and BchH. As shown in the image, BchI and BchD build a large (about 600 kDa) 2-ring complex. Subunit BchI builds up the bottom ring, while the top ring is by BchD. BchI belongs to the so-called AAA+ family of ATPases. It uses the energy of ATP to drive the Mg-chelatase reaction. It is an example of a molecular machine. In a later section, I will discuss more examples of molecular machines.
Conservation of Sequence & Structure
Significant variations in the amino acid sequences within a protein family often lead to similar three-dimensional structures. This observation suggests a higher degree of structure conservation than sequence conservation. This can be understood in the context of function: processes such as ligand binding, protein-protein interactions, and structural dynamics all rely on the three-dimensional structure. Therefore, maintaining high conservation of the three-dimensional structure is essential for function. Determining the structure of a protein with an unknown function and comparing it to known structures in a database has helped identify structural homologs, which can ultimately reveal the protein’s function. The principles of structure conservation also enable us to use structure prediction and modeling when no experimental structure is available. AI-based models by AlphaFold (Nobel Prize in Chemistry 2024) and ESM Metagenomics Atlas are widely used in protein research.