Fragment Screening & Fragment-Based Drug Discovery
Screening a fragment library can be conducted with a significantly smaller number of compounds compared to a library of drug-like molecules. An identified hit can then serve as a starting point for further optimization in a process known as fragment-based drug design (FBDD). Some details of this process are discussed in this section.
Fragment Screening & Fragment-Based Drug Discovery
In the traditional high-throughput screening method (HTS), a library of drug-like compounds, which may consist of hundreds of thousands or even millions of compounds, is screened against the protein target. Such screening may identify several active compounds that bind to the drug target and inhibit its activity. However, a more efficient method that requires a much smaller pool of compounds is fragment-based drug discovery (FBDD). In this approach, screening is conducted using a library of small molecule fragments (molecular weight not exceeding 300 Da), commonly referred to as a “fragment library.” Since the molecules used in this case are smaller and less complex than drug-like molecules, the probability of finding a molecule that will fit into the protein binding site is higher. Using fragments, it is also possible to better sample the chemical space. For choosing compounds for the library, instead of the Lipinski Rule of 5, discussed in the previous section, the Rule of Three has been suggested (Congreve et al. (2003), Drug Disc. Today, 8:876-877)
- MW 100-300
- LogP ≤ 3.0
- H-Bond Acceptors ≤ 3
- H-Bond Donors ≤ 3
- Rotatable bonds ≤ 3
- Polar Surface Area ≤ 60 Å2
Fragment-based drug design enables the compound library to remain much smaller (it could be just a few hundred rather than hundreds of thousands), making it easier to manage practically while still allowing for exploration of a broad chemical space. The goal is to identify weak yet efficient binders, characterized by high binding energy for the atoms involved. The bound fragments may subsequently be developed into more potent compounds in a process called hit expansion. An additional advantage of this approach is that it allows for more efficient control over essential properties, such as solubility, size, and toxicity of the new molecules. The aim is, of course, to identify drug-like molecules with stable binding, good solubility, and low toxicity while being economical to synthesize.
Although fragments typically display relatively low activity in assays, they can be easily screened using biophysical techniques such as NMR spectroscopy, surface plasmon resonance (SPR), weak-affinity chromatography (WAC), isothermal titration calorimetry (ITC), thermal-shift assay, or X-ray crystallography. Even in this context, co-crystallizing fragments with the protein target will significantly aid further optimization. Currently, numerous examples, ranging from identifying the first fragment hits to hit expansion, lead generation, and optimization, have progressed all the way to drug candidates and clinical trials.
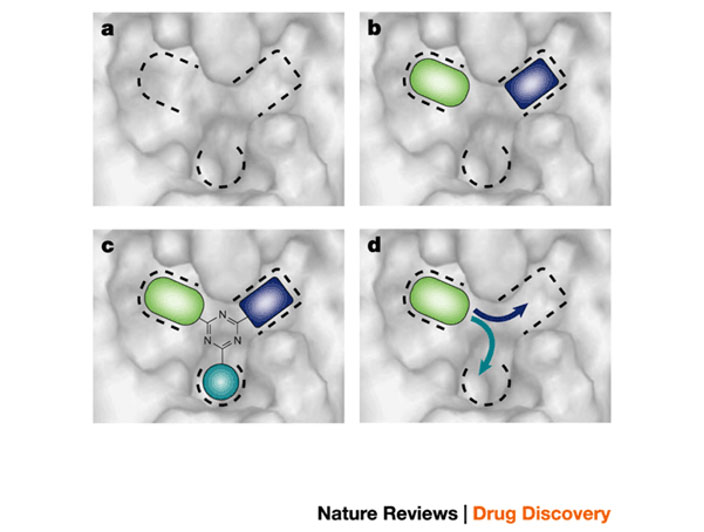
Schematic image showing how fragment-based lead discovery is applied to a case of a binding site comprising three potential binding pockets
a) Screening locates molecular fragments that bind into one (d), two (b), or all three pockets (c).
b) A lead compound is designed by organizing three fragments around a core template, as shown in (c)
c) It could be grown out from a single fragment, as shown in (d)
More details in: Blundell et al. (2002), Nature Rev Drug Discov. 1, 45-54.